OBJECTIVE—The purpose of this study was to assess the effect of glimepiride on insulin sensitivity and secretion in subjects with type 2 diabetes.
RESEARCH DESIGN AND METHODS—After a 2-week washout from prior sulfonylurea therapy, 11 obese subjects with type 2 diabetes underwent euglycemic and hyperglycemic clamp studies before and during glimepiride therapy.
RESULTS—Glimepiride resulted in a 2.4-mmol/l decrease in fasting plasma glucose (P = 0.04) that was correlated with reductions in postabsorptive endogenous glucose production (EGP) (16.4 ± 0.6 vs. 13.5 ± 0.5 μmol · kg−1 · min−1, P = 0.01) (r = 0.21, P = 0.01). Postabsorptive EGP on glimepiride was similar to that of control subjects (12.8 ± 0.9 μmol · kg−1 · min−1, NS). Fasting plasma insulin (66 ± 18 vs. 84 ± 48 pmol/l, P = 0.05), and first-phase (19 ± 8 vs. 32 ± 11 pmol/l, P = 0.04) and second-phase incremental insulin responses to glucose (48 ± 23 vs. 72 ± 32 pmol/l, P = 0.02) improved with glimepiride therapy. Insulin sensitivity did not change with treatment (4.6 ± 0.7 vs. 4.3 ± 0.7 μmol · kg−1 · min−1 · pmol−1) and remained below that of control subjects (8.1 ± 1.8 μmol · kg−1 · min−1 · pmol−1, P = 0.04).
CONCLUSIONS—The current study demonstrates that glimepiride improves both first and second phases of insulin secretion, but not insulin sensitivity, in individuals with type 2 diabetes.
Sulfonylureas have been used in the treatment of type 2 diabetes for over 40 years (1). It is generally accepted that these agents exert their hypoglycemic effect in part through direct action on the pancreatic β-cell to augment insulin secretion (1). Improvements in insulin sensitivity have also been reported in some (2,3), but not all (4), studies. Whether increased insulin sensitivity, when observed, is secondary to an amelioration of glucose toxicity with improvement in glycemic control or to a direct effect of the sulfonylureas on insulin-sensitive tissues remains unclear. Although in vitro studies have demonstrated extrapancreatic sulfonylurea effects (5,6), experiments in patients with type 1 diabetes who are incapable of increasing their insulin release have not found beneficial effects of sulfonylureas on glycemic control (7,8).
Glimepiride is the most recently approved second-generation sulfonylurea for the treatment of type 2 diabetes (9). Glimepiride differs from other sulfonylureas in that it reportedly binds to a different receptor at the β-cell membrane than other agents in this class (10). Animal studies demonstrate greater reductions in plasma glucose per increment in plasma insulin with glimepiride than with glyburide or glipizide, suggesting that glimepiride may have direct extra pancreatic effects that stimulate an improvement in insulin sensitivity (5).
The purpose of this study was to assess the effect of glimepiride on insulin sensitivity and secretion using euglycemic and hyperglycemic clamp studies in subjects with type 2 diabetes. Subjects were selected for study by having fair to good glycemic control at entry to minimize the effect of amelioration of glucose toxicity on any observed improvement in insulin sensitivity or secretion.
RESEARCH DESIGN AND METHODS
The protocol was reviewed and approved by the Institutional Review Board for Biomedical Research at the University of Pittsburgh and the General Clinical Research Center (GCRC) Review Committees. Informed, written consent was obtained from each subject before participation in the study. All studies took place in the GCRC at the University of Pittsburgh Medical Center.
Subjects
The study was comprised of eight men and three women with type 2 diabetes. The control subjects were five men and two women with normal glucose tolerance, according to National Diabetes Data Group (NDDG) standards (11) who were without a family history of type 2 diabetes. Subjects were matched for age (60 ± 8 vs. 59 ± 7 years) and BMI (30.5 ± 4.7 vs. 30.9 ± 5.6 kg/m2).
All subjects with diabetes had been receiving a stable dose of a sulfonylurea for at least 1 month before study entry. Fasting plasma glucose levels were required to be ≤8.9 mmol/l (160 mg/dl) on current therapy to be eligible. No subject was taking any medication known to interfere with glucose tolerance (i.e., thiazide diuretics, β-blockers, or steroids). Once eligibility was established, the current sulfonylurea was discontinued for a 2-week washout period. Subjects who maintained a fasting glucose level >8.3 mmol/l (150 mg/dl) and <16.6 mmol/l (300 mg/dl) were scheduled for admission to the GCRC for study.
All subjects were encouraged to maintain their usual diet and activities for the duration of the study. Subjects were asked to refrain from smoking or vigorous exercise for 2 days before each clamp procedure.
Study procedures
Subjects were admitted to the GCRC between 1800 and 1900 h the evening before the first glucose clamp studies, at which time they were fed a standard diet (60% carbohydrate, 25% fat, and 15% protein). After this meal, no food or drink other than water was permitted until completion of studies the next day.
At 0300 h of the study day, a deep antecubital vein was cannulated and a primed (30 mCi) continuous (0.3 mCi/min) infusion of [2-3H]glucose was initiated via Harvard infusion pump. At 0630 h, a dorsal hand vein was cannulated in a retrograde manner and maintained at 65°C in a Plexiglass thermoregulated hot box for sampling of arterialized venous blood throughout the procedure.
At 0700, 0730, and 0800 h basal blood samples were drawn for glucose, insulin, C-peptide, glucagon, and glucose specific activity (SA). At 0800 h, a primed continuous (1 mU · kg−1 · min−1) insulin infusion was initiated. A variable rate glucose infusion (40% dextrose) was initiated and adjusted at 5-min intervals using the glucose clamp technique to achieve target glucose concentrations of 5.5 mmol/l (100 mg/dl) for 2 h. Sampling for glucose, insulin, C-peptide, glucagon, and glucose SA was repeated at 20-min intervals. At 1000 h, the insulin and [2-3H]glucose infusions were discontinued to allow for dissipation of exogenous insulin. The glucose infusion rate (GIR) was adjusted to prevent hypoglycemia and then discontinued.
At 1100 h, an intravenous bolus infusion of 50% dextrose was administered over 1–2 min in a dose calculated to increase the plasma glucose level to 13.3 mmol/l (240 mg/dl). Plasma glucose levels were measured every 5 min and maintained at 13.3 mmol/l (240 mg/dl) via a variable rate glucose infusion as 40% dextrose. Sampling for insulin, glucagon, and C-peptide was performed at baseline, 2.5, 5, 10, 15, and 20 min following the initial glucose bolus, and then every 20 min for the duration of the study (120 min).
At the conclusion of the study, the glucose infusion was discontinued, intravenous lines were removed, and subjects were fed lunch and discharged. Diabetic subjects were instructed to start glimepiride at a dose of 2 mg per day with breakfast on the following day. Dose adjustments were made at weekly intervals based on the results of home glucose monitoring in an attempt to achieve a fasting glucose of 5–8.9 mmol/l (90–160 mg/dl).
After 4 months of therapy with the study drug, diabetic subjects were readmitted to the GCRC for repeat euglycemic and hyperglycemic clamp studies. Glimepiride was administered at 0730 h of the second study day. At the conclusion of the second study, subjects were advised to resume their previous sulfonylurea.
Analytic determinations
HbA1c concentrations were determined by high-performance liquid chromatography (Biorad, Hercules, CA). Plasma glucose concentrations were determined by the glucose oxidase method with a YSI glucose analyzer (Yellow Springs Instruments, Yellow Springs, OH). Plasma insulin, glucagon, and C-peptide were determined by radioimmunoassay (12). Plasma glucose radioactivity was determined as previously described (13).
Calculations
Rates of glucose appearance (Ra) and glucose utilization (Rd) were calculated using steady-state and non-steady-state equations defined and modified by Steele et al. (14). Endogenous glucose production (EGP) was defined as the total glucose Ra minus the GIR. First-phase insulin secretion was taken as both the maximal insulin response and the mean incremental insulin response during the first 10 min following the glucose bolus. Second-phase insulin was taken as the mean incremental insulin response during the last hour of the hyperglycemic clamp. Insulin sensitivity (Si) was calculated from the last 30 min of the euglycemic clamp as glucose Rd divided by the mean plasma insulin (90–120 min). Insulin resistance and secretion were also evaluated using the homeostasis model assessment (HOMA) (15).
Statistics
Data are mean ± SEM. In the diabetic subjects, results before and during treatment were compared with those of nondiabetic subjects. Treatment effect was estimated by calculating the difference between pre- and poststudy values for each end point. The treatment effects were summarized by means and SDs and tested for significant changes with the signed rank test and ANOVA. Comparisons between diabetic subjects at either baseline or postglimepiride to nondiabetic subjects were tested with the Wilcoxon test. Association of EGP with fasting glucose and insulin levels was examined by plotting and calculating Spearman rank correlation coefficients.
RESULTS
Dose titration
The dose of glimepiride was titrated at weekly intervals in an attempt to achieve a fasting plasma glucose of <7.8 mmol/l (140 mg/dl). The average dose of the study medication taken by the diabetic subjects at the conclusion of the study was 7.6 ± 1.7 mg/day (range 2–16 mg/day).
Fasting plasma glucose, insulin, C-peptide, and HbA1c
Fasting plasma glucose levels decreased with glimepiride therapy in the diabetic subjects (P = 0.04), but remained higher than in control subjects (P = 0.01) (Table 1). HbA1c did not change during the study period (8.0 ± 1.2 vs. 8.4 ± 1.7%). Fasting plasma insulin increased, while C-peptide (Table 1) and glucagon (85 ± 8 vs. 77 ± 8 pg/ml, NS) concentrations did not change during glimepiride treatment. Fasting insulin, C-peptide, and glucagon (nondiabetic subjects 77 ± 8 pg/ml) levels did not differ significantly from levels observed in control subjects (Table 1).
Euglycemic clamp
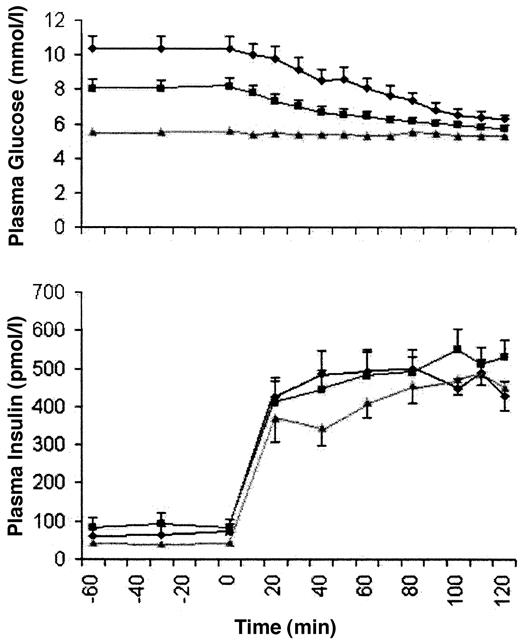
Valid tracer data were available for 10 of 11 diabetic subjects (Fig. 1 and Table 1). Plasma glucose and insulin concentrations at steady state (90–120 min) during the euglycemic clamp were similar in the diabetic subjects during each study period. Coefficients of variation for plasma glucose during the steady-state periods were similar among the diabetic groups pre- and post-treatment and the control subjects (2.9 ± 0.7, 2.2 ± 0.8, and 2.3 ± 0.4%, respectively). The GIR required to maintain euglycemia at steady state increased with treatment in the diabetic subjects and remained significantly lower than in nondiabetic subjects (Table 1).
Postabsorptive EGP and glucose Rd, which were higher in diabetic subjects before glimepiride therapy, decreased to levels that were not significantly different from nondiabetic subjects with therapy (Table 1). A significant positive correlation was observed between basal rates of EGP and fasting plasma glucose (r = 0.21, P = 0.01). EGP during steady state was suppressed to a similar extent in all three study groups (1.7 ± 2.9 vs. 2.2 ± 1.4 vs. −1.0 ± 2.0 μmol · kg−1 · min−1, P = 0.25). Rd at steady state did not improve in diabetic subjects and remained lower than that observed in nondiabetic subjects. No improvement was observed for any measure of insulin sensitivity (SI) with glimepiride therapy [Rd/IRI (90–120 min) 4.6 ± 0.7 vs. 4.3 ± 0.7; M/I 3.1 ± 1.1 vs. 3.2 ± 0.9, P = 0.73]. SI remained significantly below that observed in nondiabetic subjects (M/I 7.4 ± 1.8, P = 0.04).
Hyperglycemic clamp
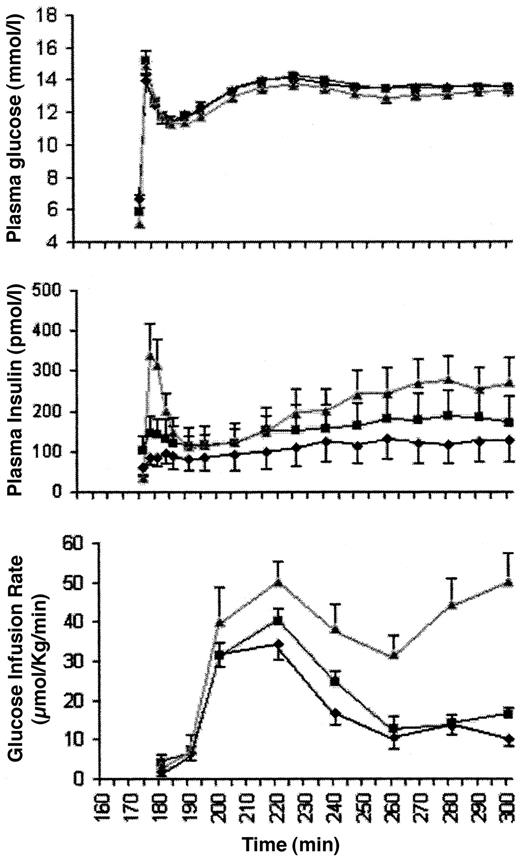
Complete insulin data were available for 10 of 11 subjects with type 2 diabetes. Steady-state plasma glucose levels during the hyperglycemic clamp study were equivalent at both study points in diabetic subjects and control subjects (Table 1), with coefficients of variation of 2.2 ± 0.3, 2.0 ± 0.3, and 1.6 ± 0.4%, respectively. Maximum (FPmax 106 ± 64 vs. 164 ± 48 pmol/l, P = 0.03) and incremental first-phase insulin secretion increased in the diabetic subjects with glimepiride (Fig.2 and Table 1). Incremental steady-state insulin also improved with glimepiride, but not to values observed in nondiabetic subjects. Mean first-phase and steady-state C-peptide responses improved with glimepiride therapy (Table 1). Plasma glucagon levels (66 ± 3 vs. 59 ± 3 pg/ml) did not change with therapy and did not differ from control subjects (59 ± 7 pg/ml).
HOMA
Results from HOMA-derived measures of insulin sensitivity in diabetic subjects before and after therapy (4.9 ± 1.3 vs. 5.5 ± 2.0) and insulin secretion (37.2 ± 12.6 vs. 63.2 ± 15.1, P < 0.01) and in control subjects (1.7 ± 0.3 and 76.9 ± 19.6, respectively) were consistent with results obtained using euglycemic and hyperglycemic clamp studies.
CONCLUSIONS
This study was initiated to assess the effect of glimepiride on insulin sensitivity and secretion in subjects with relatively well-controlled type 2 diabetes. This was done to avoid the confounding effect of amelioration of glucose toxicity as a contributor to any potential improvements in insulin action or secretion. Despite the fact that subjects were under reasonably good control on prior sulfonylurea therapy at study entry, a further improvement in fasting glucose levels was observed as measured by a reduction in fasting plasma glucose concentration with glimepiride (Table 1). This effect of glimepiride was associated with an improvement in β-cell function as assessed by an increase in fasting and incremental first- and second-phase insulin responses during hyperglycemic clamp experiments (Table 1). However, no improvement in insulin sensitivity was observed using euglycemic clamp experiments and HOMA.
The observed improvement in first-phase insulin release with glimepiride during a hyperglycemic clamp in individuals with type 2 diabetes was unexpected since sulfonylureas are generally thought to only affect second-phase insulin release (1). An augmentation in first-phase insulin release has been reported with two first generation sulfonylureas. In one study, an increase in first-phase insulin secretion was observed following an acute intravenous bolus of tolbutamide, a short-acting sulfonylurea, in untreated hyperglycemic (blood glucose 12 ± 0.5 mmol/l) subjects with type 2 diabetes (16). When glucose levels were decreased to 8 ± 1 mmol/l in these subjects, no effect on first-phase insulin secretion was observed. Augmentation of first-phase insulin secretion was also observed with gliclazide, which is not available in the U.S. (17). In one prior report using the second-generation sulfonylurea glipizide, early insulin release was reported as being normalized after a mixed meal in a group of individuals with type 2 diabetes independent of dietary control (18). However, only summed and area under the curve insulin concentrations following the mixed meal were reported as significant, and results were not further fractionated into time points to quantify first- or second-phase insulin secretion.
The hyperglycemic clamp is accepted as a rigorous method for determining both first and second phases of insulin secretion (19). Disturbances in β-cell function, characterized by reduced or absent acute insulin responses to intravenous glucose, are observed early in the course of type 2 diabetes (20), as well as in first-degree relatives at risk for type 2 diabetes (21). These changes in early insulin secretion are considered to be important in the pathogenesis of type 2 diabetes (22). Positive correlations have been observed between first-phase insulin secretion and insulin responses during oral glucose tolerance testing (23), suggesting that early abnormalities in β-cell function are predictive of deterioration to abnormal glucose tolerance.
In a previous hyperglycemic clamp study of the second-generation sulfonylureas glipizide and glyburide, an increase in both first- and late-phase insulin secretion was observed in nondiabetic individuals and only in second-phase insulin secretion in those with type 2 diabetes (24,25). Clark and Matthews (26) reported improvements in second-phase insulin release after 1 week of treatment with glimepiride or glibenclamide during a hyperglycemic clamp study in subjects with type 2 diabetes when compared with placebo. Changes in first-phase insulin secretion were not addressed.
The study of Clark and Matthews differed from the current study in that improvements in insulin sensitivity during euglycemic clamp studies were also observed with each of the sulfonylurea agents when compared with placebo (26). There are important differences in study design that may have contributed to this difference for measures of insulin sensitivity. Subjects in this previous study were withdrawn from a prior sulfonylurea for only 1 week before each 7-day study period with glimepiride, glibenclamide, or placebo. In addition, treatments were given in random order with no washout phase between study periods. Those treated with placebo were significantly more hyperglycemic, suggesting that the improvement in insulin sensitivity may have been due to an amelioration of glucose toxicity (27). In addition, euglycemic clamps in the study by Clark and Matthews targeted a whole blood glucose level of 3.5 mmol/l for 3 h, which is different from the target of 5.5 mmol/l for 2 h in the current study.
Improvements in insulin sensitivity are reported in prior studies investigating extrapancreatic mechanisms of action of the sulfonylureas. However, in these studies, subjects were under poor glycemic control at baseline with fasting glucose levels in excess of 11.1 mmol (>200 mg/dl) (2,3,28), thus making it difficult to attribute the cause of an increased insulin sensitivity to the sulfonylurea or to the amelioration of glucose toxicity.
The observed improvement in fasting glucose in diabetic subjects was due, at least in part, to observed reductions in EGP since postabsorptive rates of glucose disposal were not improved. However, the most likely explanation for this was the increase in insulin. Reductions in glucagon concentrations have been proposed as another mechanism by which sulfonylureas reduce fasting hyperglycemia; however, no significant decrease in plasma glucagon was observed in this study (1). It is also possible that a reduction in glucose toxicity contributed, but subjects were in relatively good glycemic control at baseline and no improvement in insulin sensitivity was observed (27).
A limitation to the current study was the lack of a control group using an alternative form of therapy to improve glucose concentrations. As glucose toxicity can impair insulin secretion as well as sensitivity, it is possible that the observed improvements in insulin secretion in this study may have been due to the 2.4-mmol/l reduction in fasting glucose. Another limitation to this study is the fact that direct comparisons were not made with other sulfonylurea agents that may have similar effects on insulin secretion if studied using similar conditions and subjects.
In conclusion, the hypoglycemic effect of glimepiride can be attributed to an increase in fasting and first and second phases of insulin secretion in individuals with type 2 diabetes. As reductions in first-phase insulin secretion are described in individuals at risk for type 2 diabetes, clinical trials of therapy targeting improvements in early insulin release as a means of preventing deterioration to type 2 diabetes may be warranted.
Article Information
This work was supported by a research grant from Aventis and funds received from the NIH/NCRR/GCRC grant nos. 5M01RR00056 and NIDDKAM20411. The authors would like to thank the subjects who volunteered to participate in this study, the nursing staff of the GCRC for their excellent management of patients, and Lisa Sinay for her careful manuscript preparation.
References
Address correspondence and reprint requests to Mary Korytkowski, Falk, Room 588, 3601 Fifth Ave., Pittsburgh, PA 15213. E-mail: korytkowski@msx.dept-med.pitt.edu.
Received for publication 2 April 2002 and accepted in revised form 12 June 2002.
M.K. is on the Speaker’s Bureau for Aventis and is a coinvestigator on an educational grant from Aventis to the University of Pittsburgh Center for Continuing Education. J.G. has received honoraria for speaking engagements, consulting fees, and research support from Novartis, Novo Nordisk, GlaxoSmithKline, Aventis, Bristol-Myers Squibb, and Pfizer, as well as consulting fees from Emisphere Pharmaceuticals.
A table elsewhere in this issue shows conventional and Système International (SI) units and conversion factors for many substances.